


PROPIEDADES DE LOS AMINOACIDOS
Reactividad química
Los aminoácidos reaccionan fácilmente debido a la naturaleza química de su radical, influyendo en la estabilidad, la reactividad y otras propiedades de las proteínas. Los derivados de hidrocarburos aromáticos como la alanina, la valina, la leucina y la isoleucina, son inertes y casi no intervienen en las reacciones químicas; los aminoácidos con grupos aminos libres son nucleofílicos como alanina, arginina y la lisina, por lo que favorecen cambios como el de oscurecimiento o bien la formación de enlaces entrecruzados.
En la histidina, el imidazol que contiene se rompe por acción de algunas sustancias, dando subproductos, que se degradan por otras rutas. El tioéter de la metionina puede sufrir oxidación-reducción, o bien el guanidino de la arginina puede ser alterado por diversos mecanismo. El sulfidrilo de la cisteína, es el más reactivo de todos los grupos ‚Ò‚ de los aminoácidos, y produce cambios deseables e indeseables. La asparrágina y la glutamina se hidrolizan fácilmente por ácidos y por álcalis, transformándose en ácido aspártico y ácido glutámico, respectivamente.
Propiedades ácido-base
Algunas propiedades se deben a su naturaleza iónica anfotérica ácido-base. A éstos compuestos también se les llama anfolitos (proviene de electrolitos anfóteros). La estructura iónica se ha establecido por estudios tales como, sus puntos de fusión que son elevados (200°C), o por su solubilidad en agua (son más solubles en agua que en disolventes polares). Tienen constantes dieléctricas elevadas, así como momentos dipolares debido a la presencia de cargas negativas y positivas dentro de la misma molécula.
Por los grupos ionizables, carboxilo, amino y otros, los aminoácidos se comportan con carga (+) o (-) según el pH en que se encuentren; esto es debido a su naturaleza anfotérica, por lo que pueden donar y recibir electrones, por lo tanto producen un estado químico llamado punto isoeléctrico (pI) o de doble ion, tienen el mismo número de cargas positivas como negativas, siendo su cargan neta cero. Los aminoácidos pueden tener tres estados que dependen del pH:
a) a pH <>
b) a pH = pI: con carga cero; y,
c) a pH > pI: con carga negativa o aniónica.
En cualquiera de los tres estados pueden atraer iones de carga contraria por fuerzas electrostáticas débiles.

La ionización de los aminoácidos es similar a la de cualquier otra molécula, y por lo tanto sigue la ecuación de Henderson-Hasselbalch:

donde pK, por definición, es el logaritmo negativo de la constante de disociación (K) del grupo ionizable:

Así como los grupos carboxilos y aminos, influyen en el comportamiento acido-base de los aminoácidos, también los hacen el imidazol de la histidina, el amino e de la lisina, el carboxilo b del ácido aspártico, el sulfidrilo de la cisteína el carboxilo del ácido glutámico, el guanidino de la arginina y el hidroxilo fenólico de la tirosina.
El punto isoeléctrico de los compuestos que contienen solo dos grupos ionizables, un amino y un carboxilo, se puede calcular a partir de sus respectivos valores de pK:

ESTRUCTURA Y NOMBRE DE AMINOÁCIDOS Y AMINAS DE INTERÉS
AMINOÁCIDOS
Estructura
Como su nombre lo indica los aminoácidos son compuestos que poseen un grupo amino (-NH2) y un grupo ácido (carboxílico -COOH) en su estructura. Los aminoácidos son los precursores de los péptidos y las proteínas, y en ellos el grupo amino y el grupo carboxilo, se encuentran unidos al mismo átomo de carbono, conocido como carbono-a (a-aminoácidos). La estructura general de los a-aminoácidos (a excepción de la prolina, que es cíclica) se muestra en la siguiente figura.

Estructura química de un aminoácido
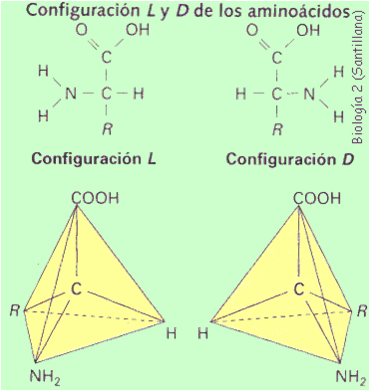
Estructura química en el plano y estructura espacial
Clasificación
Como se puede apreciar, el carbono-a (a excepción de la glicina) es un carbono quiral y como tal presenta dos enantiómeros (L- y D-).
Los 20 a-aminoácidos presentes en las proteínas son de la serie L- en su representación de Fischer poseen el grupo amino hacia la izquierda. La diferencia entre los eeeeeedada por el resto -R, o cadena lateral, unida al carbono-alfa.
Atendiendo a la naturaleza del grupo -R los aas. pueden clasificarse en:
• Neutros o apolares
• Polares sin carga
• Polares con carga negativa
• Polares con carga positiva
Neutros o Apolares.
Son 8 los aminoácidos que se clasifican como poseedores de cadenas laterales no polares. La alanina, valina, leucina e isoleucina, poseen cadenas laterales de hidrocarburos alifáticos. La metionina posee una cadena lateral de éter tiólico (C-S-C). La prolina es el único aminoácido cíclico, pues el grupo -R se cierra sobre el N del grupo a-amino (realmente es un amina secundaria). Por su parte, la fenilalanina y el triptófano contienen grupos aromáticos.


Polares sin carga
Siete son los a-aminoácidos cuyo resto -R es polar pero sin carga. La glicina posee la cadena más simple, un átomo de hidrógeno. La serina y la treonina son portadores de un grupo hidroxilo (-OH). La asparragina y la glutamina, poseen cadenas laterales portadoras de un grupo amida, y por hidrólisis dan lugar, respectivamente, a aspartato y glutamato, dos aminoácidos con carga negativa. La tirosina posee un grupo fenólico y la cisteína debe su polaridad a la presencia de un grupo tiólico (-SH).

Polares con carga negativa.
Existen dos a-aminoácidos cuyo resto polar posee carga negativa a pH fisiológico, debida a la presencia de un grupo carboxilo (-COOH) , el ácido glutámico y el ácido aspártico.

Polares con carga positiva. Tres son los a-aminoácidos que poseen restos -R cargados positivamente a pH fisiológico. La lisina posee una cadena lateral de butilamonio, la arginina presenta un grupo -R de guanidina y la histidina es portadora de un grupo -R de imidazolio.

Esta clasificación se ha realizado en base al grupo -R, pero es importante indicar que a pH fisiológico (pH 7,3), el grupo a-amino se encuentra cargado positivamente y el grupo a-carboxilo lo está negativamente.
Dentro del conjunto de los aminoácidos naturales, existen unos que pueden ser sintetizados por las células humanas a partir de otras sustancias, pero también hay aminoácidos que debemos tomarlos en la dieta, ya que nuestras células no pueden sintetizarlos o, cuando menos, no en cantidad suficiente para satisfacer la demanda del organismo; se conocen con el nombre de aminoácidos esenciales y son valina, leucina, isoleucina, treonina, metionina, fenilalanina, triptófano y lisina.
http://www.um.es/molecula/prot.htm
http://es.wikipedia.org/wiki/Amino%C3%A1cido
http://es.encarta.msn.com/encyclopedia_761555775/Amino%C3%A1cidos.html
http://www.biologia.edu.ar/macromoleculas/aminoaci.htm
http://biomodel.uah.es/model3/aa.htm
PROTEÍNAS
En diferentes áreas médicas y biológicas, es importante el conocimiento de las propiedades de las proteínas. Son macromoléculas con pesos moleculares entre miles y millones, y constituyen la maquinaria de la vida; cientos de proteínas han sido identificadas como enzimas, otras son proteínas estructurales, otras realizan funciones diversas como las proteínas contráctiles, las de almacenamiento‚ las de transporte, etc.
Composición y tamaño de las proteínas:
Las proteínas son moléculas muy complejas, en cuya composición elemental se encuentran siempre presentes carbono, hidrógeno, oxígeno y nitrógeno aunque la mayoría de ellas incluye al azufre y en algunas se sabe de la presencia de fósforo, hierro, zinc, molibdeno y otros elementos.
Estructuralmente, los elementos mencionados se encuentran distribuidos en unidades estructurales llamadas aminoácidos, que unidos entre sí forman estructuras poliméricas o polipéptidos; las proteínas se consideran fundamentalmente polímeros de aminoácidos.
De acuerdo con la composición química las proteínas, se clasifican en dos tipos principales:
A) SIMPLES:
Están constituidas únicamente por L–a–aminoácidos o sus derivados y comprenden los siguientes grupos:
1. - Albúminas.
2. - Globulinas.
3. - Glutelinas.
4. - Prolaminas.
5. - Albuminoides.
6. - Histonas.
7. - Protaminas.
B) CONJUGADAS:
Tienen en su composición otras moléculas diferentes además de los aminoácidos los cuales se llaman grupos prostéticos, unidas por fuerzas distintas a las atracciones iónicas; están los siguientes grupos:
1. Nucleoproteínas.
2. Glucoproteínas o mucoproteínas.
3. Fosfoproteínas.
4. Cromoproteínas.
5. Lipoproteínas.
6. Metaloproteínas.
Si consideramos que la unión de las proteínas establece un patrón determinado, donde cada péptido contiene un grupo terminal de carboxilo libre en el extremo derecho de la cadena y un grupo terminal amino libre en el extremo izquierdo, como ya vimos, al unirse el carboxilo libre y el amino libre se forma un polipéptido.

Debido a éstas características se pueden clasificar en dos clases principales sobre la base de su estructura y solubilidad; éstas son:
I.- Proteínas fibrosas:
A) Proteínas Fibrosas Simples: debido a su estructura molecular, parecida a una fibra, son muy insolubles en los solventes comunes, como agua, solución salina diluida, solventes orgánicos, ácidos y álcalis diluidos. También se les conoce como albuminoides o escleroproteínas. Ejemplos: queratina, colágeno y elastina.
B) Proteínas Fibrosas Conjugadas: realmente se conoce poco de éste tipo, pero tal vez el pigmento de las plumas de las aves pertenezca a este tipo.
II.- Proteínas globulares:
A) Proteínas Globulares Simples: Son solubles en agua, solución salina diluida, solventes orgánicos, ácidos y álcalis diluidos. Se dividen a su vez en dos grupos; solubles en agua destilada, y en insolubles en agua destilada:
1) Proteínas globulares simples solubles en agua destilada:
a) Albúminas: Proteínas muy solubles, que pueden precipitarse de una solución acuosa por saturación con una sal ácida como el (NH4)2SO4, o por saturación con una sal neutra como Na2SO4, se coagulan por calentamiento. Ejemplo: lactalbúmina y albúminas del suero.
b) Seudoglobulinas: son proteínas solubles que pueden precipitarse de una solución acuosa por saturación de uno a tres cuartos con una sal ácida como sulfato de amonio.
c) Protaminas: muy solubles, son polipéptidos básicos en estado natural, no se coagulan por calentamiento, en su estructura predominan los aminoácidos básicos; precipitan a otras proteínas. Se encuentran principalmente en las células huevo. Ejemplo: la salamina (del salmón) y la esturina (del esturión).
d) Histonas: solubles, son básicas, precipitan por adición de hidróxido de amonio NH4OH; coagulan por el calor. Ejemplo: nucleohistonas de los núcleos.
2) Proteínas globulares simples insolubles en agua:
a) Euglobulinas: Insolubles en agua destilada, pero solubles en soluciones diluidas de sal, pueden precipitarse de una solución salina diluida por semisaturación con una sal ácida como el sulfato de amonio, o por saturación con una sal neutra como el sulfato de sodio; se coagulan por calentamiento. Ejemplo: seroglobulina y ovoglobulina.
b) Prolaminas: Insolubles en agua destilada, solubles en álcalis diluidos y en soluciones alcohólicas al 60% y 80%; insolubles en alcohol absoluto. Ejemplo: la zeína del maíz y la gliadina del trigo.
c) Glutelinas: Insolubles en agua destilada, en soluciones alcohólicas y solventes neutros, pero solubles en álcalis diluidos, coagulan por el calor. Ejemplo: Glutelina del trigo.
B) Proteínas Globulares Conjugadas: Se encuentran en la naturaleza combinadas con no proteínas: se dividen en varias clases, dependiendo del grupo prostético:
1) Cromoproteínas: Proteínas combinadas con pigmentos. Ejemplo: hemoglobina, hemocianina, citocromo y flavoproteínas.
2) Glucoproteínas: Proteínas combinadas con carbohidratos; por hidrólisis dan aminoazúcares (hexosaminas). Ejemplo: la proteína mucina y las proteínas del plasma.
3) Lipoproteínas: Proteínas combinadas con grasas neutras (triglicéridos) o con otros lípidos como fosfolípidos y colesterol.
4) Fosfoproteínas: Proteínas combinadas con ácido fosfórico, o un radical que contiene fósforo que no sea un fosfolípido ni ácido nucleico. Ejemplo: caseína.
5) Nucleoproteínas: Formadas por una o varias moléculas de proteínas unidas a una clase única de ácido nucleico. Ejemplo: nucleína, nucleohistona extraída de tejidos que contienen muchos núcleos (como los tejidos glandulares).
6) Metaloproteínas: Proteína combinada con metales, como el cobre (ceruplasmina) o el hierro (siderofilina).
Las proteínas varían en su tamaño y complejidad; hay algunas muy grandes y complejas, como el sistema enzimático que participa en la conversión del ácido pirúvico en acetil-Co-A que tiene un peso molecular de 10 millones e incluye varios grupos prostéticos distintos; la insulina por otra parte está formada por sólo 51 aminoácidos y su peso molecular es de 5,800. Hay polímeros con pesos moleculares menores que el de la insulina, como algunas hormonas y antibióticos; sin embargo, por convención, sólo se consideran como proteínas aquellos polímeros de aminoácidos con pesos moleculares semejantes o mayores al peso molecular de la insulina.
PROPIEDADES DE LAS PROTEÍNAS:
Las proteínas debido a que están compuestas totalmente o en su mayor parte por aminoácidos, poseen propiedades que reflejan en gran medida las propiedades de estos constituyentes.
Propiedades ácido-base de las proteínas en solución
Punto isoeléctrico:
Aunque la mayoría de los grupos carboxilo y grupos amínicos de los aminoácidos se bloquean cuando éstos se unen para formar las uniones peptídicas, siempre quedan libres algunos de éstos grupos, ya sea en los extremos de las cadenas polipeptídicas, o en las cadenas laterales de los aminoácidos acídicos y básicos. La disociación de los grupos ionizables que están presentes en las proteínas, ocurre como en el caso de los grupos ionizables de los aminoácidos individuales, se gobierna por el pH del medio en el que se encuentra la proteína. A pH de 7.0 o en valores cercanos a ésta condición, que son los habituales en la mayoría de las células, los grupos carboxilo de los ácidos aspártico y glutámico se encuentran en sus formas básicas cargadas negativamente, mientras que la lisina y arginina están presentes en sus formas acídicas, cargadas positivamente. La carga total de la molécula proteica, depende pues, del pH de la solución y del número relativo de cada aminoácido en la molécula. Así, cuando el pH de la solución es tal que la carga neta de la molécula proteica es cero, es decir, cuando el número total de cargas negativas iguala al número total de cargas positivas presentes en la molécula, se llama a éste valor de pH, punto isoeléctrico o pH isoeléctrico de la proteína. A continuación enumeramos los valores del punto (o pH) isoeléctrico (pI) de varias proteínas. Como en el caso individual de cada aminoácido, las proteínas pueden adquirir carga neta durante su titulación.
CLASIFICACION DE LAS PROTEÍNAS
De acuerdo con la composición química las proteínas, se clasifican en dos tipos principales:
A) SIMPLES:
Están constituidas únicamente por L–a–aminoácidos o sus derivados y comprenden los siguientes grupos:
1. - Albúminas.
2. - Globulinas.
3. - Glutelinas.
4. - Prolaminas.
5. - Albuminoides.
6. - Histonas.
7. - Protaminas.
B) CONJUGADAS:
Que tienen en su composición otras moléculas diferentes además de los aminoácidos los cuales se llaman grupos prostéticos, unidas por fuerzas distintas a las atracciones iónicas; están los siguientes grupos:
1. Nucleoproteínas.
2. Glucoproteínas o mucoproteínas.
3. Fosfoproteínas.
4. Cromoproteínas.
5. Lipoproteínas.
6. Metaloproteínas.
Si consideramos que la unión de las proteínas establece un patrón determinado, donde cada péptido contiene un grupo terminal de carboxilo libre en el extremo derecho de la cadena y un grupo terminal amino libre en el extremo izquierdo, como veremos más adelante, al unirse el carboxilo libre y el amino libre se forma un polipéptido; debido a éstas características se pueden clasificar en dos clases principales sobre la base de su estructura y solubilidad; éstas son:
I.- Proteínas fibrosas:
A) Proteínas Fibrosas Simples: debido a su estructura molecular, parecida a una fibra, son muy insolubles en los solventes comunes, como agua, solución salina diluida, solventes orgánicos, ácidos y álcalis diluidos. También se les conoce como albuminoides o escleroproteínas. Ejemplos: queratina, colágeno y elastina.
B) Proteínas Fibrosas Conjugadas: realmente se conoce poco de éste tipo, pero tal vez el pigmento de las plumas de las aves pertenezca a este tipo.
II.- Proteínas globulares:
A) Proteínas Globulares Simples: Son solubles en agua, solución salina diluida, solventes orgánicos, ácidos y álcalis diluidos. Se dividen a su vez en dos grupos; solubles en agua destilada, y en insolubles en agua destilada:
1) Proteínas globulares simples solubles en agua destilada:
a) Albúminas: Proteínas muy solubles, que pueden precipitarse de una solución acuosa por saturación con una sal ácida como el (NH4)2SO4, o por saturación con una sal neutra como Na2SO4, se coagulan por calentamiento. Ejemplo: lactalbúmina y albúminas del suero.
b) Seudoglobulinas: son proteínas solubles que pueden precipitarse de una solución acuosa por saturación de uno a tres cuartos con una sal ácida como sulfato de amonio.
c) Protaminas: muy solubles, son polipéptidos básicos en estado natural, no se coagulan por calentamiento, en su estructura predominan los aminoácidos básicos; precipitan a otras proteínas. Se encuentran principalmente en las células huevo. Ejemplo: la salamina (del salmón) y la esturina (del esturión).
d) Histonas: solubles, son básicas, precipitan por adición de hidróxido de amonio NH4OH; coagulan por el calor. Ejemplo: nucleohistonas de los núcleos.
2) Proteínas globulares simples insolubles en agua:
a) Euglobulinas: Insolubles en agua destilada, pero solubles en soluciones diluidas de sal, pueden precipitarse de una solución salina diluida por semisaturación con una sal ácida como el sulfato de amonio, o por saturación con una sal neutra como el sulfato de sodio; se coagulan por calentamiento. Ejemplo: seroglobulina y ovoglobulina.
b) Prolaminas: Insolubles en agua destilada, solubles en álcalis diluidos y en soluciones alcohólicas al 60% y 80%; insolubles en alcohol absoluto. Ejemplo: la zeína del maíz y la gliadina del trigo.
c) Glutelinas: Insolubles en agua destilada, en soluciones alcohólicas y solventes neutros, pero solubles en álcalis diluidos, coagulan por el calor. Ejemplo: Glutelina del trigo.
B) Proteínas Globulares Conjugadas: Se encuentran en la naturaleza combinadas con no proteínas: se dividen en varias clases, dependiendo del grupo prostético:
1) Cromoproteínas: Proteínas combinadas con pigmentos. Ejemplo: hemoglobina, hemocianina, citocromo y flavoproteínas.
2) Glucoproteínas: Proteínas combinadas con carbohidratos; por hidrólisis dan aminoazúcares (hexosaminas). Ejemplo: la proteína mucina y las proteínas del plasma.
3) Lipoproteínas: Proteínas combinadas con grasas neutras (triglicéridos) o con otros lípidos como fosfolípidos y colesterol.
4) Fosfoproteínas: Proteínas combinadas con ácido fosfórico, o un radical que contiene fósforo que no sea un fosfolípido ni ácido nucleico. Ejemplo: caseína.
5) Nucleoproteínas: Formadas por una o varias moléculas de proteínas unidas a una clase única de ácido nucleico. Ejemplo: nucleína, nucleohistona extraída de tejidos que contienen muchos núcleos (como los tejidos glandulares).
6) Metaloproteínas: Proteína combinada con metales, como el cobre (ceruplasmina) o el hierro (siderofilina).
Las proteínas varían en su tamaño y complejidad; hay algunas muy grandes y complejas, como el sistema enzimático que participa en la conversión del ácido pirúvico en acetil-Co-A que tiene un peso molecular de 10 millones e incluye varios grupos prostéticos distinto; la insulina por otra parte está formada por sólo 51 aminoácidos y su peso molecular es de 5,800. Hay polímeros con pesos moleculares menores que el de la insulina, como algunas hormonas y antibióticos; sin embargo, por convención, sólo se consideran como proteínas aquellos polímeros de aminoácidos con pesos moleculares semejantes o mayores al peso molecular de la insulina.
LAS PROTEINAS EN EL METABOLISMO
Metabolismo de las Proteínas
En el metabolismo, el principal producto final de las proteínas es el amoníaco (NH3) que luego se convierte en urea (NH2)2CO2 en el hígado y se excreta a través de la orina.
Trataremos primordialmente de la transformación catabólica del nitrógeno de los aminoácidos en urea y de los esqueletos de carbono en intermediarios anfibólicos del ciclo del ácido cítrico.
CATABOLISMO DEL NITRÓGENO DE LOS AMINOÁCIDOS
En los tejidos de mamíferos los grupos amígenos de los aminoácidos, derivados ya sean de la dieta o de la demolición de las proteínas tisulares son excretada, en último término, como urea en la orina. La biosíntesis de la urea implica la acción de varias enzimas. Se puede dividir convenientemente para su estudio en 4 procesos:
1 Transaminación,
2 Desaminación oxidativa,
3 Transporte de amoniaco, y
4 Reacciones del ciclo de laurea.
La relación de estas áreas con el catabolismo global del nitrógeno de los aminoácidos se muestra en la Fig. 2-8.

Fig. 2.8. Flujo global del nitrógeno en el catabolismo de los aminoácidos
Otros vertebrados distintos de los mamíferos comparten todos los caracteres de este esquema, exceptuando la síntesis de urea. La urea, el producto final característicos del metabolismo del nitrógenos de los aminoácidos en el hombre y otros animales ureotélicos, es reemplazada por el ácido úrico en los organismos uricotélicos (reptiles y aves) o por amoniaco en los organismos amonotélicos (por ejemplo los teleósteos).
Cada uno de los 4 procesos será considerado ahora en detalle. Aunque todos desempeñan un papel en la biosíntesis de los aminoácidos, lo que sigue se estudia desde el punto de vista del catabolismo de los aminoácidos.
Transaminación
La transaminación catalizada por las enzimas llamadas transaminasas o aminotransferasas, implica la interconver-sión de un par de aminoácidos y un par de cetoácidos. Estos generalmente son a–amino y b–cetoácidos (Fig. 2-9).

El fosfato de piridoxal, la vitamina B6 en forma de coenzima, forma una parte esencial del sitio activo de las transaminasas y de muchas otras enzimas con aminoácidos como sustratos. En todas las reacciones de los aminoácidos, dependientes del fosfato de piridoxal, el paso inicial es la formación de una base de Shiff intermediaria unida a la enzima. Fig. 2-10.

Este intermediario estabilizado por la reacción recíproca con una región catiónica del sitio activo, se puede estructurar de maneras que incluyen la liberación de un cetoácido con formación de fosfato de piridoxamina unido a la enzima. La forma unida, aminada de la coenzima puede formar entonces una base análoga intermediaria de Shiff con un cetoácido. Durante la transaminación, la coenzima unida sirve, así como un transportador intermediario de grupos amígenos. Fig. 2-10. Puesto que la constante de equilibrio para la mayor parte de las reacciones de transaminasas es cercana a la unidad, la transaminación es un proceso libremente reversible. Esta reversibilidad permite a las transaminasas funcionar tanto en el catabolismo de los aminoácidos como en su biosíntesis.

Dos transaminasas, la alanina–pirúvico transaminasa (alanintransaminasa) y la glutámico–a–cetoglutárico transaminasa (glutámico–transaminasa), presentes en la mayor parte de los tejidos animales, catalizan la transferencia de grupos amígenos de la mayor parte de los aminoácidos para formar alanina (a partir del piruvato) o del glutamato (a partir del a–cetoglutarato).
Cada transaminasa es específica para el par especificado de aminoácido y cetoácido como un par de sustratos, pero inespecífica para el otro par, el cual puede ser cualquiera de una amplia variedad de aminoácidos y sus correspondientes cetoácidos. Puesto que la alanina es también un sustrato para la reacción de la glutámico transaminasa, todo el nitrógeno amínico proveniente de los aminoácidos que pueden experimentar la transaminación se puede concentrar en el glutamato. Esto es importante porque el L–glutamato es el único aminoácido de los tejidos de mamífero que experimenta desaminación oxidativa a una tasa apreciable. La formación de amoniaco de los grupos amígenos a se realiza, así, principalmente mediante la conversión del nitrógeno amínico a del L–glutamato.
La mayor parte de los aminoácidos (pero no todos) son sustratos de la transaminación. Las excepciones incluyen a la lisina, la treonina y a los iminoácidos cíclicos prolina e hidroxiprolina. La transaminación no está restringida a los grupos amígenos a. El grupo amígeno d de la ornitina es, por ejemplo, fácilmente transaminado formando g–semialdehído del glutamato (Fig. 2-11).
Desaminación oxidativa
La conversión oxidativa de muchos aminoácidos en sus correspondientes a–cetoácidos ocurre en homogeneizados de tejidos hepáticos y renal de mamíferos. Aunque la mayor parte de la actividad de los homogeneizados frente a los L–a–aminoácidos se debe a la acción conjunta de las transaminasas y de la L–glutámico deshidrogenasa, tanto la actividad de la L- como la D–aminoacidooxidasa se presentan en los tejidos hepáticos y renal de los mamíferos y están ampliamente distribuidas en otros animales y en los microorganismos. Se debe notar, sin embargo, que no se conoce el papel fisiológico de la L- y de la D–aminoácido-oxidasa en los tejidos de mamífero.
Las aminoácido-oxidasas son flavoproteínas autooxidables, es decir, el FMN o el FAD reducido es reoxidado directamente por el oxígeno molecular (Fig. 2–12), formando el peróxido de hidrógeno (H2O2) sin participación de los citocromos o de otros transportadores de electrones. El producto tóxico H2O2 es desdoblado entonces en O2 y H2O por la catalasa que existe ampliamente en los tejidos especialmente en el hepático. (Fig. 2–12). Aunque las reacciones de las aminoácido-oxidasas son reversibles, el a–cetoácido producido no es descarboxilado enzimáticamente por el H2O2 si falta la catalasa, formándose un ácido carboxílico con un átomo menos de carbono. Tanto la actividad de la L- como la de la D–aminoácido-oxidasa están presentes en el tejido renal, aunque la función de la D–aminoácido-oxidasa es oscura.

En las reacciones de la aminoácido-oxidasa (Fig. 2–12) el aminoácido es deshidrogenado primero por la flavoproteína de la oxidasa, formando un a–iminoácido. Este adiciona agua espontáneamente y luego se descompone espontáneamente en el correspondiente a–cetoácido con pérdida del nitrógeno a–imínico como amoniaco.
La L-aminoácido-oxidasa de los mamíferos una FMN–flavoproteína, está restringida a los tejidos renal y hepático. Su actividad es bastante es bastante baja y esencialmente no tiene efecto sobre la glicina o sobre los L–isómeros de los ácidos dicarboxílicos o de los b–hidroxi–a-aminoácidos. Así, no es verosímil que esta enzima desempeñe un papel principal en el catabolismo de los aminoácidos en los mamíferos.
La D-aminoácido-oxidasa de los mamíferos una FAD–flavoproteína de amplia especificidad de substratos, existe en el tejido hepático y renal de la mayor parte de los mamíferos. La D–asparagina y la D–glutamina no son oxidadas y la glicina y los D–isómeros de los aminoácidos ácidos y básicos son malos substratos. La significación fisiológica de esta enzima en los mamíferos se desconoce.
L–Glutámico deshidrogenasa. Los grupos amígeno de la mayor parte de los aminoácidos son transferidos, en último término, al a–cetoglutarato por transaminación formando L–glutamato (Fig. 2-8). La liberación de este nitrógeno como amoniaco es catalizada por la L–glutámico deshidrogenasa, una enzima de gran actividad, ampliamente distribuida en los tejidos de mamíferos (Fig. 2-13). La glutámico deshidrogenasa hepática es una enzima regulada cuya actividad es afectada por modificadores alostéricos como el ATP, el GTP y el NADP, que inhiben a la enzima, y el ADP que la activa. Ciertas hormonas también parecen influir sobre la actividad de la glutámico deshidrogenasa. La glutámico deshidro-genasa usa ya sea NAD+ o NADP+ como cosubstrato. La reacción es reversible y funciona tanto en el catabolismo de los aminoácidos como en su biosíntesis. Por consiguiente, ella funciona no sólo canalizando el nitrógeno del glutamato hacia urea (catabolismo), sino también catalizando la aminación del a–cetoglutarato por el amoniaco libre. Esta última función (biosintética) es de particular importancia en las plantas y en las bacterias, las cuales pueden sintetizar grandes cantidades de aminoácidos a partir de la glucosa y el amoniaco. Cuando el ganado bovino es alimentado con dietas ricas en carbohidratos y nitrógeno en la forma de urea, las bacterias del rumen convierten primero a la urea en amoniaco, luego utilizan la reacción de la glutámico deshidrogenasa proporcionando al ganado una dieta abundante en glutamato y otros aminoácidos.
Además del amoniaco formado en los tejidos, una considerable cantidad es producida por las bacterias intestinales, tanto a partir de las proteínas de la dieta, como de la urea presente en los líquidos secretados en el aparato digestivo. Este amoniaco es absorbido en el intestino y pasa a la sangre de la vena porta, la cual característicamente contienen concentra-ciones mayores de amoniaco que la sangre de la circulación general. En circunstancias normales, el hígado prontamente elimina el amoniaco de la sangre de la vena porta, de manera que la sangre que abandona el hígado (y, de hecho, toda la sangre periférica) está virtualmente exenta de amoniaco. Esto es esencial, ya que aun diminutas cantidades de amoniaco son tóxicas para el sistema nervioso central. Los síntomas por intoxicación por amoniaco incluyen un temblor peculiar en aleteo, lenguaje farfullado, visión borrosa y, en los casos graves, coma y muerte. Estos síntomas se parecen a los del síndrome del coma hepático. Por lo tanto, el tratamiento incluye medidas encaminadas a reducir los niveles sanguíneos de amoniaco.

Cuando la función hepática está gravemente menoscabada o cuando se establecen comunicaciones colaterales entre la vena porta y las venas de la circulación general (como puede ocurrir en la cirrosis), la sangre porta puede evadir al hígado. El amoniaco proveniente de los intestinos puede así, elevarse a niveles tóxicos en la sangre de la circulación general. Los procedimientos de derivación quirúrgica (fístula de Eck u otras formas de derivación portacava) también conducen a la intoxicación por amoniaco, particularmente después de la ingestión de grandes cantidades de proteínas o de hemorragia del aparato digestivo. El contenido de amoniaco de la sangre que abandona los riñones por la vía de las venas renales siempre excede al de las arterias renales, indicando que los riñones producen amoniaco y los vierten a la sangre. Sin embargo, la excreción en la orina del amoniaco producido por las células de los túbulos renales constituye un aspecto mucho más importante del metabolismo renal del amoniaco. La producción del amoniaco forma parte de los mecanismos de los túbulos renales que regulan el equilibrio ácido-básico, así como de conservación de los cationes. La producción de amoniaco por los riñones está marcadamente aumentada en la acidosis metabólica y deprimida en la alcalosis. No sólo se deriva de la urea, sino también de los aminoácidos intracelulares, particularmente de la glutamina. La liberación de amoniaco es catalizada por la glutaminasa renal (Fig. 2-14).

Transporte de amoniaco
Aunque el amoniaco puede ser excretado como sales de amonio —particularmente en estado de acidosis metabólica— la vasta mayoría es excretada como urea, el principal componente nitrogenado de la orina. El amoniaco producido constantemente en los tejidos por los procesos descritos anteriormente, sólo se encuentra como vestigios de la sangre (10 – 20 mg/100 ml) puesto que es rápidamente eliminado de la circulación por el hígado y convertido, ya sea en glutamato, glutamina o urea. Estos niveles vestigiales de amoniaco contrastan claramente con las cantidades más considerables de aminoácidos libres, particularmente de glutamina, en la sangre (cuadro 15-1)
La eliminación del amoniaco mediante la reacción de la glutámico deshidrogenasa fue mencionada anteriormente. La formación de glutamina es catalizada por la glutamina sintetasa (Fig. 2-15), una enzima mitocondrial presente en máxima cantidad en el tejido renal. La síntesis del enlace amídico de la glutamina se lleva a cabo a expensas de la hidrólisis de un equivalente de ATP en ATP y Pi. La reacción es así fuertemente favorecida en la dirección de la síntesis de glutamina.

La liberación del nitrógeno amídico de la glutamina como amoniaco sucede no por reversión de la reacción de la glutamina sintetasa, sino por formación hidrolítica de amoniaco catalizada por la glutaminasa (Fig. 2-14). La reacción de la glutaminasa, a diferencia de la reacción de la glutamina sintetasa, no incluye la participación de nucleótidos de adenina, favorece fuertemente la formación de glutamato y no funciona en la síntesis del mismo. Estas 2 enzimas, la glutamina sintetasa y la glutaminasa (Fig. 2-16) sirven para catalizar la interconversión del ion amonio libre y la glutamina de una manera que recuerda la interconversión de la glucosa, y la glucosa–6–fosfato por la glucocinasa y la glucosa–6– fosfatasa (Fig. 2-5). Una reacción análoga es catalizada por la L–asparaginasa de origen animal, vegetal y microbiano. La asparaginasa y la glutaminasa han sido empleadas, ambas, como agentes antitumorales ya que ciertos tumores tienen requerimientos anormalmente elevados de glutamina y asparagina.

Mientras que en el encéfalo el mecanismo principal para la eliminación del amoniaco es la formación de glutamina, en el hígado la vía más importante es la formación de urea. El tejido del encéfalo puede formar urea, aunque esto no desempeña un papel significativo en la eliminación del amoniaco. En el encéfalo, la formación de glutamina debe ser precedida por la síntesis de glutamato en el propio encéfalo porque el aporte de glutamato sanguíneo es inadecuado para explicar las cantidades aumentadas de glutamina formadas dentro del encéfalo en presencia de niveles altos de amoniaco sanguíneo. La fuente inmediata de glutamato para este propósito es el a–cetoglutarato. Esto empobrecería rápidamente el aporte de intermediarios del ciclo del ácido cítrico a menos que pudieran ser repuestos por la fijación de CO2 con la conversión del piruvato en oxalacetato. El efecto, en el encéfalo sucede una fijación importante de CO2 en forma de aminoácidos, presumiblemente por la vía del ácido cítrico y después de la infusión de amoniaco más oxalacetato es desviado hacia la síntesis de glutamina (en vez de aspartato) por la vía del a–cetoglutarato.
Regulación del ciclo de la urea (síntesis de la urea)
Un hombre moderadamente activo que consuma cerca de 300 g de carbohidratos, 100 g de grasa y 100 g de proteínas diariamente debe excretar aproximadamente 16.5 g de nitrógeno al día. 95% es eliminado por los riñones el 5% restante, en su mayor parte como nitrógeno en las heces. La principal ruta para la excreción de nitrógeno en el hombre es la de la urea sintetizada en el hígado, vertida a la sangre y eliminada por el riñón. En el hombre que se alimenta con una dieta occidental, la urea constituye el 80 – 90 % del nitrógeno excretado.
Las reacciones y los intermediarios en la biosíntesis de 1 mola de urea a partir de 1 mola de amoniaco y otra de bióxido de carbono (activados con Mg++ y ATP), así como del nitrógeno a–amínico del aspartato se muestran en la figura 2-17. El proceso global requiere de 3 molas de ATP (dos de las cuales son convertidas en ADP + Pi y una en AMP y Ppi) y la participación sucesiva de 5 enzimas que catalizan las reacciones numeradas de la Fig. 2-17. De los 6 aminoácidos que intervienen en la síntesis de la urea, uno, el N–acetilglutamato, funciona como un activador enzimático y no como un intermediario. Los 5 restantes —asparta-to, arginina, ornitina, citrulina y arginin-succinato— funcionan todos como transportadores de átomos que en último término se vuelven urea. Dos de ellos (aspartato y arginina) existen en las proteínas, mientras que los tres restantes (ornitina, citrulina y argininsuccinato) no. El principal papel metabólico de éstos tres últimos aminoácidos es, en los mamíferos, la síntesis de urea. Nótese que la formación de urea es, en parte un proceso cíclico. La ornitina usada en la reacción 2 es regenerada en la reacción 5. Así, no hay pérdida ni ganancia neta de ornitina, citrulina, argininsuccinato o de arginina durante la síntesis de la urea; sin embargo, el amoniaco, el CO2, el ATP y el aspartato si son consumidos.
Reacción 1: síntesis del carbamoilfosfato.
La condensación de una mola de amoniaco, de otra de bióxido de carbono y de una de fosfato (derivada del ATP) para formar carbamoilfosfato es catalizada por la carbamoilfosfato sintetasa, una enzima presente en las mitocondrias hepáticas de todos los organismos ureotélicos, incluyendo al hombre. Las 2 molas de ATP hidrolizadas durante esta reacción aportan la fuerza quimiomotriz para la síntesis de dos enlaces covalentes del carbamoil-fosfato: el enlace amídico y el enlace del anhídrido mixto ácido carboxílico –ácido fosfórico. Además de Mg++ se requiere de un ácido dicarboxílico, de preferencia N-acetilglutamato. El papel exacto del N–acetilglutamato no se conoce con certeza. Su presencia lleva a cabo un profundo cambio conformacional en la estructura de la carbamoilfosfato sintetasa que expone a ciertos grupos sulfhidrilo, oculta a otros y afecta la afinidad de la enzima por el ATP.
En las bacterias, la glutamina sirve como sustrato, en lugar del amoniaco, para la síntesis del carbamoilfosfato. Una reacción semejante catalizada por la carbamatocinasa es también importante en la utilización de la citrulina por las bacterias.
Reacción 2: Síntesis de citrulina.
La transferencia de la fracción carbamoílo del carbamoilfosfato a la ornitina, formando citrulina + Pi, es cata-lizada por la L–ornitintranscarbamoilasa de las mitocondrias del hígado. La reacción es altamente específica para la ornitina y el equilibrio favorece grandemente la síntesis de la citrulina.
Reacción 3: Síntesis del argininsuccinato.
En la reacción de la arginin-succinato sintetasa, el aspartato y la citrulina son unidos mediante el grupo amígeno del aspartato. La reacción requiere de ATP y el equilibrio favorece fuertemente la síntesis de arginin-succinato.
Reacción 4: Desdoblamiento del argininsuccinato en arginina y fumarato.
El desdoblamiento reversible del argininsuccinato en arginina y fumarato es catalizado por la argininsuccinasa, una enzima friolábil de los tejidos hepático y renal de los mamíferos. La pérdida de actividad en el frío se acompaña de la disociación en 2 componentes proteínicos. Esta disociación es impedida por el Pi, la arginina y el argininsuccinato o por el p–hidroximercuribenzoato, el cual no tiene efecto adverso sobre la actividad. La reacción se lleva a cabo por un mecanismo de trans-eliminación. El fumarato formado puede ser convertido en oxalacetato mediante las reacciones de la fumarasa y de la malicodeshidrogenasa (Fig. 2-5) y luego transaminado éste para regenerar el aspartato.
Reacción 5: Desdoblamiento de la arginina en ornitina y urea.Esta reacción completa el ciclo de la urea y regenera la ornitina, sustrato de la reacción 2. El desdoblamiento hidro-lítico del grupo guanídico de la arginina es catalizado por la arginasa, la cual se encuentra en el hígado de todos los organismos ureotélicos. Cantidades meno-res de arginasa también existen en el tejido renal, en el encéfalo, en la glándula mamaria, en el tejido testicular y en la piel. La arginasa altamente purificada preparada en el hígado de mamífero es activada por el Co++ o el Mn++. La ornitina y la lisina son potentes inhibidores que compiten con la arginasa.
No hay comentarios:
Publicar un comentario